Projects
Genomics of Hodgkin and Non-Hodgkin Lymphoma
Background
Although Hodgkin lymphoma (HL) is relatively uncommon (~7000–7500 new cases diagnosed annually in the United States), it comprises ~10% of lymphomas in the western world. The treatment of HL is relatively successful. However, patients who relapse or are refractory to treatment have few secondary treatment options, and overall survival remains low in this patient population.
Objective
A better understanding of the pathobiology of this disease is an important clinical goal. This project aims to address these challenges by using cutting-edge genomic technologies to study Hodgkin lymphoma, which is defined by a rare malignant cell population. Traditional methods struggle with such cancers, leaving many aspects of its biology poorly understood.
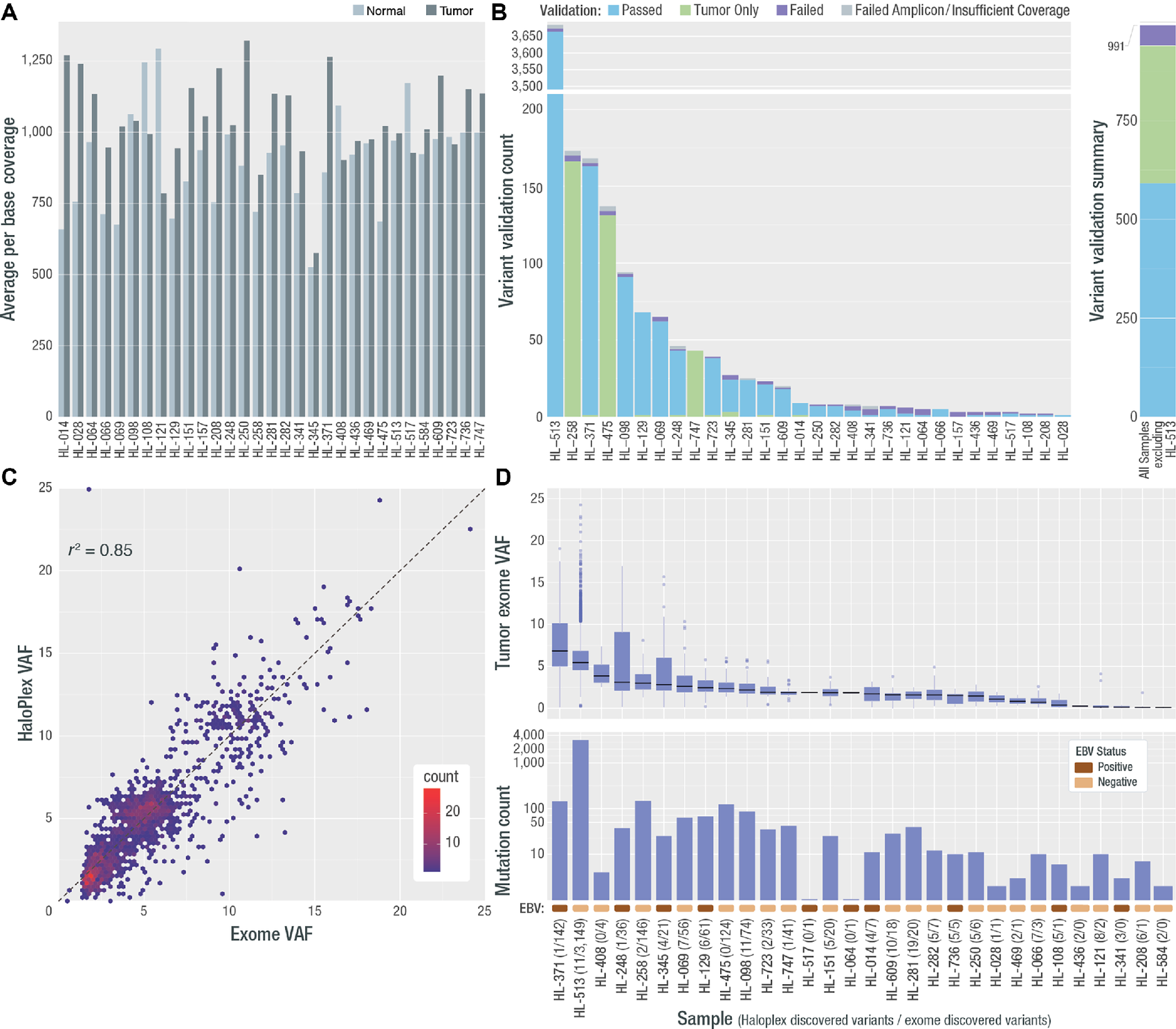
Deep exome coverage, validation rate, final variant count, and VAF per sample. A, Average per base coverage for targeted regions across each tumor and normal sample. B, A targeted orthogonal sequencing strategy (HaloPlex; Agilent) was used to validate all variants that passed filtering strategies for all samples (except for the hypermutated sample, for which a subset of variants were selected for validation—see Materials and Methods). The count of assayed variants that passed, failed, or were validated only in the HaloPlex tumor sample, is shown by sample as well as the number for which validation was not possible due to low tumor coverage or amplicon design failure. The overall count of variants that passed, failed, or received a “tumor only” validation status, excluding variants from HL-513 is shown at the right. Note: One patient (HL-584) did not have any variants pass filtering and review, therefore 30/31 patients were included in validation. C, Comparison of exome VAF and HaloPlex VAF is shown for samples with two or more variants. D, VAF and variant count for all variants across all samples used in all further analyses. Following each sample number is the number of HaloPlex discovered variants and exome discovered variants. In addition, a colored rectangle indicating the EBV stats (assessed using competitive alignment) is shown. Note: Variants from one patient failed validation (HL-157) and no further variants were called in de novo exercises. This patient was removed from all further analyses. In addition, the patient who did not have variants to validate, gained two variants in the de novo exercise and was included in the final cohort (HL-584).
Health Disparities in Multiple Myeloma
Background
Multiple myeloma (MM) is a malignant proliferation of monoclonal plasma cells in bone marrow that accounts for 14% of all hematologic cancers and is essentially incurable. It is accepted that all MM is preceded by the benign expansion of clonal plasma cells called monoclonal gammopathy of undetermined significance (MGUS) or smoldering MM (SMM). When self-identified race is used, African Americans (AAs) have a twofold increase in risk for MM compared to European Americans (EAs) and are diagnosed at younger ages. Consistent with the increase in risk, AAs have a fourfold higher prevalence of MGUS, and MGUS often occurs at younger ages in AA patients.
Objective
Our research aims to elucidate the genomic, transcriptomic, and tumor microenvironment (TME) differences between multiple myeloma (MM) patients of European (EA) and African American (AA) descent that may explain some of the health disparities associated with this disease. Currently we are collaborating with Dr. Linda Baugh (Mayo Clinic Rochester) to study the tumor microenvironment in AA and EA MM patients.
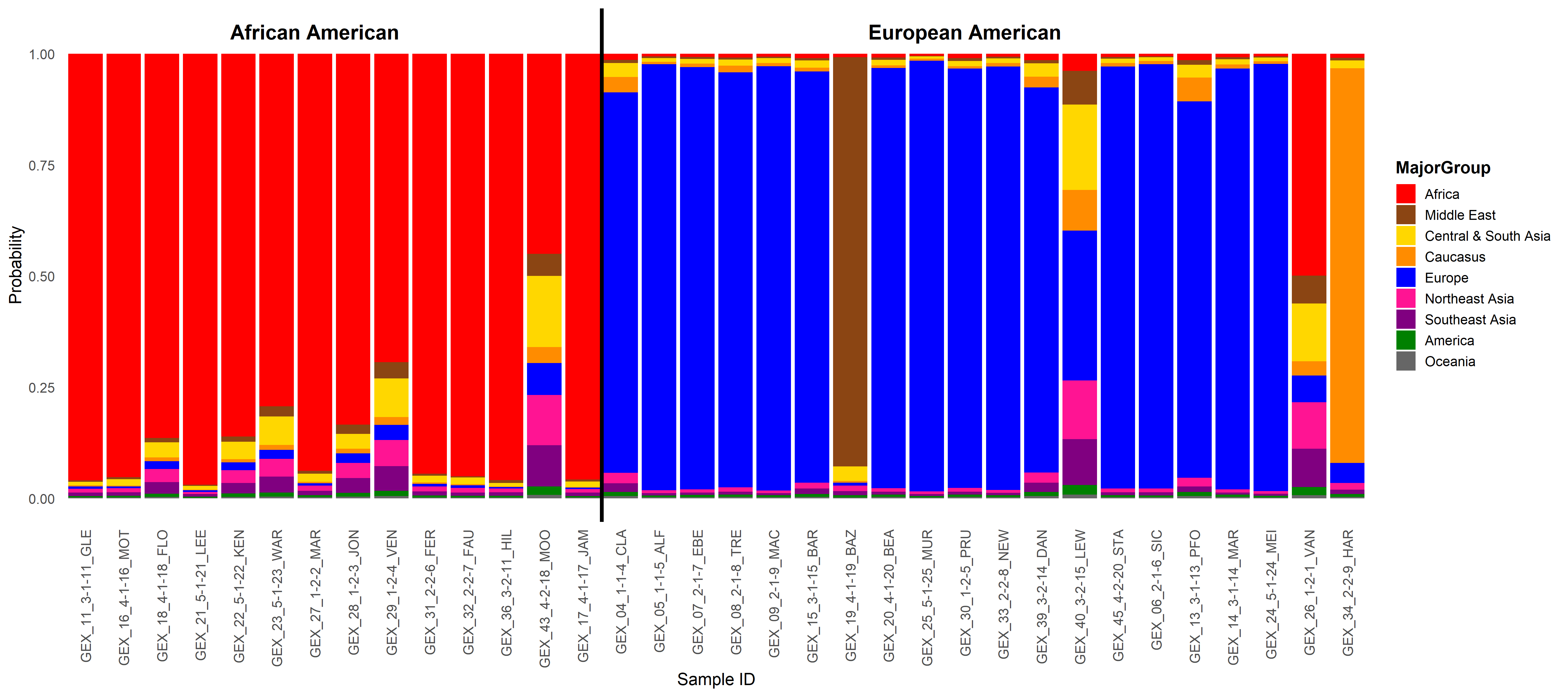
Ancestry admixture proportion was calculated for each patient. The probability of ancestry from each of nine major geographical ancestral groups was determined using a support vector machine (SVM) with the HGDP as the reference panel. The percentage of ancestry from each global population for each patient is shown on the y-axis. The patients are grouped by their self-reported race.